笔者之前写过一些转录组分析工具的使用方法,有比较流行的,也有最近才发表的,这一期为大家带来的是2011年发表的目前引用量高达3800+的包含联配(调用bowtie/bowtie2/star)、组装定量打包分析的工具RSEM。为什么要写这一篇已经出来7年的工具,两个原因,一是笔者最近在做了众多权衡之后换成RSEM做日常的分析,二是最近生信草堂要出一本推文集,这也是这篇推文写作的价值之一。
文章连接地址如下:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-323
安装RSEM
我们首先进入rsem的github地址:https://github.com/deweylab/RSEM ,在 Clone or download区找到下载地址,开始下载。笔者在不同时间测试了下下载速度,可能由于国内对github做了限制的原因,下载速度比较慢,翻墙后下载速度正常。
make #编译
make ebseq #编译ebseq,ebseq为rsem官方推荐的差异基因分析工具,该工具发表于2013年,目前引用次数为500+
make install #安装
使用RSEM
RSEM有两种分析方法,一是从fasta格式开始进行分析直到得到表达矩阵,二是用现有的bam文件进行下一步分析,下面的流程是第一种方法的有参流程,分建库、表达定量和合并成矩阵三步;
1.建库:
rsem可以调用第三方软件bowtie2、star等进行分析,但是不能直接使用这些软件自身建立的索引,需要使用rsem-prepare-reference针对bowtie2和star分别建立索引,方法如下:
rsem-prepare-reference --gtf Homo_sapiens.GRCh38.89_chr_version.gtf --star hg38.fa ./human_ensembl
这里需要提供gtf注释文件和fa基因组文件,整体上参数比较简单。因为笔者是要做转录组分析,所以选择了准确度更好一点的STAR作为内部调用工具,所以加上参数--star。
2.表达量计算:
针对单个样本:
rsem-calculate-expression --paired-end -p 20 --star --append-names --star-output-genome-bam /data/rawdata/rawdata-first/C01/trim/C01_combined_R1_val_1.fq /data/ rawdata/rawdata-first/C01/trim/C01_combined_R2_val_2.fq /data /genome/human_ensemble_chr/rsem-star/human_ensembl ./C01
--paired-end #针对双端数据
-p #线程,越多越好
--star #内部调用STAR做联配
--append-names #将gene或者tran的name和ID用“_”连接起来,这个参数可选可不选
--star-output-genome-bam #输出正常STAR工具标准输出的bam文件
这一步因为需要调用STAR,对内存有一定的要求;如果是人的基因组建库,则服务器内存最好大于32GB。
3.合并成表达矩阵
rsem官方提供了将多个样本合并成一个表达矩阵的工具,使用如下:
rsem-generate-data-matrix ./C01/C01.genes.results ./C02/C02.genes.results . /C03/C03.genes.results >output_matrix.txt
结果截图如下:
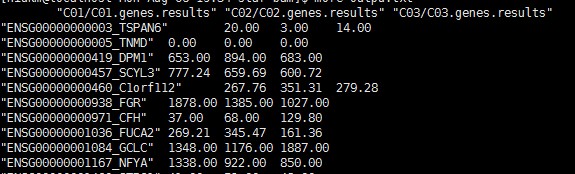
笔者并不喜欢官方提供的脚本,并且只有count矩阵结果,没有TPM和FPKM矩阵,所以可以写一个简单的python脚本重新将所需要的结果提取出来。
vim rsem-count-extract.py
#!/usr/bin/env python
#coding: utf-8
import sys
from itertools import islice
mydict = {}
for a in sys.argv[1:]:
c = open(a, 'r')
for line in islice(c, 1, None):
a = line.strip().split()
key = a[0]
value = a[4]
#可以选择其他列
if key in mydict:
mydict[key] = mydict[key] + '\t' + value
else:
mydict[key] = value
for key,value in mydict.items():
print(key + '\t' + value)
然后将python脚本的调用整合在shell脚本里,
list_csv=""
list_name="gene"
for i in ./C*/*genes.results
do
echo $i
list_csv=${list_csv}" "${i}
i_name1=${i%/*}
i_name2=${i_name1##*/}
list_name=${list_name}" "${i_name2}
echo ${list_name}
done
echo $list_name > ./gene-count-matrix.txt
python ./rsem-count-extract.py ${list_csv} >> ./gene-count-matrix.txt
这样就可以批量提取到样本名称正确的count矩阵,同时如果想求TPM或者FPKM的矩阵,可以修改python脚本中value = a[4]的数值为5或者6。
得到表达矩阵后,后续分析可以采用官方推荐的ebseq或者比较常用的deseq2做差异基因分析。对于转录本的差异分析目前来说并不建议,这个主要还是因为现有算法对转录本定量的精确度并不高。
从整体上看,RSEM还是很全面的,首先它由于调用了STAR做联配,所以效率高速度快,其次结果中count、TPM、FPKM都有,对后续差异分析和WGCNA分析提供了便利,通过STAR的中间bam文件使用stringtie又可以做lncRNA分析,其针对转录本独特的算法也是相对提高了转录本定量的精确度。所以从整体上来说,笔者认为RSEM在目前的同类软件中相对来说是比较完美的了,但不一定是最好的。展望未来,年青一代的孩子编程能力越来越强,新的算法和高用户体验性的工具也许正在路上。
转自生信草堂公众号,已授权
生信草堂
浙大生信博士团队倾力打造的一个科研人员学习交流的公众微信平台。我们致力于科研社区服务,分享前沿的科技进展,提供生信分析方法,解读经典分析案例,公众数据库的挖掘和临床数据统计分析。在此我们欢迎各位的加入!加微信bioinformatics88拉您进生信交流群返回搜狐,查看更多

